- Сломанный ген что это значит у ребенка
- Патогенез синдрома ломкой Х-хромосомы
- Фенотип и развитие синдрома ломкой Х-хромосомы
- Лечение синдрома ломкой Х-хромосомы
- Риски наследования синдрома ломкой Х-хромосомы
- Генетические нарушения у человека и методы их выявления
- Методы исследования хромосом
- Мутации в генах и заболевания, к которым они способны приводить
- Как выявляют рецессивные мутации?
- Что делать, если в семье есть наследственное заболевание?
Сломанный ген что это значит у ребенка
Этиология и встречаемость синдрома ломкой Х-хромосомы. Синдром ломкой Х-хромосомы (MIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене FMR1 в Xq27.3. Синдром ломкой Х-хромосомы встречается с частотой 16-25 на 100 000 в общей популяции среди мужчин и в два раза реже среди женщин.
Синдром ломкой Х-хромосомы составляет 3-6% всех случаев умственной отсталости среди мальчиков с положительным семейным анамнезом по умственной отсталости при отсутствии врожденных пороков.
Патогенез синдрома ломкой Х-хромосомы
Продукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может сопровождать определенный подкласс мРНК от ядра к рибосомам.
Более 99% мутаций в гене FMR1 — экспансия нуклеотидного повтора (CGG)n в 5′-нетранслируемом участке гена. В нормальных аллелях FMR1 число повторов CGG составляет от 6 до приблизительно 50. В патогенных аллелях (или при полных мутациях) количество повторов более 200. Аллели с более чем 200 повторами CGG обычно имеют гиперметилированную последовательность повторов CGG и смежного промотора FMR1. Гиперметилирование инактивирует промотор FMR1, вызывая снижение экспрессии FMRP.
Полные мутации возникают из аллелей премутации (от 59 до 200 повторов CGG) с передачей мутантного аллеля FMR1 от матери (но не от отца); фактически при отцовской передаче премутации часто, наоборот, сокращаются. Полные мутации не могут возникать из нормальных аллелей. Поскольку длина неустойчивых повторов CGG увеличивается в каждом последующем поколении, если они передаются женщиной, обычно наблюдается увеличение числа пораженных потомков в последующих поколениях в семье; этот феномен называется генетической антиципацией.
Риск экспансии премутации в полную мутацию возрастает с увеличением числа повторов в премутации. Тем не менее не все премутации одинаково предрасположены к экспансии. Хотя премутации встречаются сравнительно часто, переход в полную мутацию наблюдают только в ограниченном количестве гаплотипов, т.е. когда есть склонность гаплотипа к экспансии.
Эта склонность гаплотипа частично может быть связана с присутствием нескольких триплетов AGG, вставленных в последовательность повторов CGG; оказывается, такие триплеты AGG тормозят экспансию повторов CGG, следовательно, их отсутствие в некоторых гаплотипах может предрасполагать к экспансии.
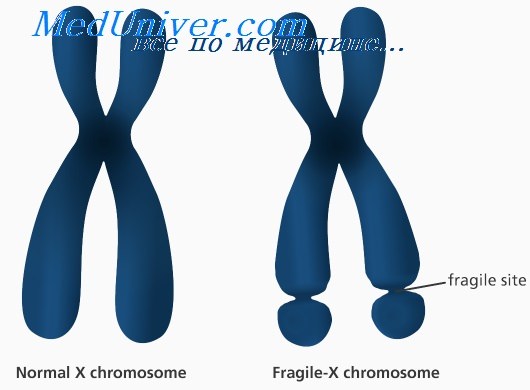
Фенотип и развитие синдрома ломкой Х-хромосомы
Синдром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее пораженные индивидуумы также имеют поведенческие аномалии, включая гиперактивность, размахивание руками, истерики, плохой зрительный контакт и признаки аутизма. Физические характеристики мужчин изменяются с пубертатом.
До полового созревания пораженные мальчики имеют несколько увеличенный размер головы и некоторые другие неотчетливые симптомы; после наступления половой зрелости у них частые более отчетливые признаки (длинное лицо с выдающейся челюстью и лбом, крупные ушные раковины, макроорхидизм).
Поскольку эти клинические признаки не уникальны для синдрома ломкой Х-хромосомы, диагноз зависит от молекулярного обнаружения мутаций. Пациенты с синдромом ломкой Х-хромосомы имеют нормальную продолжительность жизни.
Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа зависит от мозаицизма метилирования повторов и их числа. Поскольку полные мутации неустойчивы, некоторые пациенты имеют смесь клеток с числом повторов, колеблющимся от премутации до полной мутации (мозаицизм числа повторов).
Все мужчины с мозаицизмом числа повторов больны, но часто имеют более высокие показатели умственного развития, чем пациенты с полной мутацией в каждой клетке; у женщин с мозаицизмом числа повторов клинические проявления варьируют от нормы до полного проявления. Аналогично некоторые пациенты имеют смесь клеток с метилированием повторов CGG и без него (мозаицизм метилирования повторов). Все мужчины с мозаицизмом метилирования больны, но часто имеют более высокие показатели умственного развития, чем с гиперметилированием в каждой клетке; женщины с мозаицизмом метилирования также могут быть здоровыми или больными.

Очень редко пациенты имеют полную мутацию, неметилированную во всех клетках; независимо от пола, степень тяжести у них варьирует от нормы до полной клиники. Кроме того, у женщин фенотип зависит от степени смещения инактивации Х-хромосомы.
Носительницы премутации (но не полных мутаций) имеют 20% риск ранней дисфункции яичников. Мужчины-носители премутации имеют риск развития синдрома FXTAS. FXTAS проявляет себя как поздняя прогрессирующая мозжечковая атаксия с интенционным тремором. У больных могут также присутствовать снижение краткосрочной памяти и двигательных функций, когнитивные нарушения, а также паркинсонизм, периферическая нейропатия, проксимальная мышечная слабость нижних конечности и дизавтономия.
Пенетрантность FXTAS зависит от возраста, обнаруживается в 17% в течение шестого десятилетия жизни, в 38% в течение седьмого десятилетия, в 47% в течение восьмого десятилетия и в трех четвертях старше 80 лет. FXTAS может встречаться и у некоторых женщин — носительниц премутации.
Особенности фенотипических проявлений синдрома ломкой Х-хромосомы:
• Возраст начала: детство
• Умственная недостаточность
• Дисморфическое лицо
• Постпубертатное увеличение яичек у мужчин (макроорхидизм)
Лечение синдрома ломкой Х-хромосомы
К настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и фармакологическое лечение поведенческих проблем.
Риски наследования синдрома ломкой Х-хромосомы
Риск того, что женщина с премутацией будет иметь больного ребенка, определяется размером премутации, полом плода и семейным анамнезом. Эмпирически риск для носителя перестройки иметь больного ребенка может достигать 50% для каждого мальчика и 25% для каждой девочки, но зависит от размера премутации. На основе анализа сравнительно небольшого количества матерей-носительниц известно, что риск повторения может снижаться, если премутация уменьшается со 100 до 59 повторов. Пренатальная диагностика доступна за счет использования ДНК плода из ворсин хориона или амниоцитов.
Пример синдрома ломкой Х-хромосомы. Р.Л., 7-летний мальчик, направлен в клинику педиатрии в связи с умственной задержкой и гиперактивностью. Он не смог посещать детский сад, поскольку был агрессивным, не в состоянии выполнять задания, имел бедные речевые и двигательные навыки. Несмотря на задержанное развитие, он не потерял основных этапов: сидел к 10-11 мес, ходить начал в 20 мес, говорил два или три ясных слова в 24 мес.
В остальном ребенок здоров. Его мать и тетя по матери имели небольшие проблемы обучения в детстве, дядя по матери умственно задержан. Данные медицинского осмотра в норме, за исключением гиперактивности.
Врач рекомендовал несколько тестов, включая кариотипирование, функциональные исследования щитовидной железы и ДНК-анализ на синдром ломкой Х-хромосомы. Анализ гена FMR1 методом блот-гибридизации по Саузерну соответствовал синдрому ломкой Х-хромосомы.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Генетические нарушения у человека и методы их выявления

Генами называются участки ДНК, в которых закодирована структура всех белков в теле человека или любого другого живого организма. В биологии действует правило: «один ген – один белок», то есть в каждом гене содержится информация только об одном определенном белке.
В 1990 году большая группа ученых из разных стран начала проект под названием «Геном человека». Он завершился в 2003 году и помог установить, что человеческий геном содержит 20–25 тысяч генов. Каждый ген представлен двумя копиями, которые кодируют один и тот же белок, но могут немного различаться. Большинство генов одинаковые у всех людей – различается всего 1%.
ДНК находится в клетке внутри ядра. Она особым образом организована в виде хромосом – эти нитеподобные структуры можно рассмотреть в микроскоп с достаточно большим увеличением. Внутри хромосомы ДНК намотана на белки – гистоны. Когда гены неактивны, они расположены очень компактно, а во время считывания генетического материала молекула ДНК расплетается.
В клетках человека есть структуры, которые называются митохондриями. Они выполняют роль «электростанций» и отвечают за дыхание. Это единственные клеточные органеллы, у которых есть собственная ДНК. И в ней тоже могут возникать нарушения. 
Весь набор хромосом в клетке называется кариотипом. В норме у человека он представлен 23 парами хромосом, всего их 46. Выделяют два вида хромосом:
- 22 пары аутосом одинаковы у мужчин и женщин. В каждой паре хромосомы имеют одинаковую длину и содержат одинаковые наборы генов.
- Одна пара половых хромосом. У женщин это две X-хромосомы. Одна из них неактивна и плотно свернута – ее называют тельцем Барра. У мужчин одна половая хромосома представлена X-хромосомой, а вторая – Y-хромосомой, она меньше по размерам.
Методы исследования хромосом
Для исследования кариотипа применяют специальный метод – световую микроскопию дифференциально окрашенных метафазных хромосом культивированных лимфоцитов периферической крови.
Этот анализ применяется для диагностики различных хромосомных заболеваний. Он позволяет выявлять такие нарушения, как:
- Грубые изменения в кариотипе – изменение количества хромосом. Например, при синдроме Дауна в клетках ребенка присутствует лишняя хромосома №21.
- Присутствие в организме клеток с разными кариотипами. Это явление называется мозаицизмом.
- Хромосомные аберрации – нарушение структуры хромосом, внутрихромосомные и межхромосомные перестройки. Сюда относятся делеции (утрата участка хромосомы), дупликации (удвоение участка хромосомы), инверсии (поворот участка хромосомы на 180 градусов), транслокации (перенос участка одной хромосомы в другую).

Однако с помощью исследования кариотипа можно выявить не все генетические нарушения. Оно не способно обнаружить такие изменения, как:
- микроделеции и микродупликации, когда утрачивается или дублируется очень маленький участок хромосомы;
- болезни обмена, вызванные нарушением последовательности «букв» генетического кода в отдельных генах;
- митохондриальные заболевания, связанные с нарушениями в генетическом материале митохондрий;
- низкопроцентный мозаицизм, когда клеток с неправильным кариотипом очень мало;
- мутации в отдельных генах, которые не приводят к изменению внешнего вида хромосом;
- эпигенетические расстройства, при которых структура хромосом и генов не меняется, но изменяется их функция.

Для получения дополнительной информации, не видимой в световой микроскоп, используют хромосомный микроматричный анализ (ХМА). С его помощью можно изучить все клинически значимые участки генома и выявить изменения в количестве и структуре хромосом, а именно микрополомки (микроделеции и микродупликации).
Во время хромосомного микроматричного анализа применяют технологию полногеномной амплификации и гибридизации фрагментов опытной ДНК с олигонуклеотидами, нанесенными на микроматрицу. Если объяснять простыми словами, то сначала ДНК, которую необходимо изучить, копируют, чтобы увеличить ее количество, а затем смешивают ее со специальными ДНК-микрочипами, которые помогают выявлять различные нарушения.
Эта методика позволяет в одном исследовании выявлять делеции и дупликации участков ДНК по всему геному. Разрешающая способность стандартного ХМА от 100 000 пар нуклеотидов – «букв» генетического кода (в отдельных регионах от 10 000 п. н.).
С помощью ХМА можно выявлять:
- изменения числа хромосом;
- дупликации и делеции, в том числе микродупликации и микроделеции;
- отсутствие гетерозиготности – утрату одной из двух копий гена. Это явление имеет важное значение в онкологии, при болезнях импринтинга (когда активность гена зависит от того, от какого из родителей он получен), аутосомно-рецессивных заболеваниях (связанных с рецессивными генами – о них мы поговорим ниже), близкородственных браках;
- однородительские дисомии, когда в геноме ребенка присутствуют две хромосомы от одного родителя.
Однако, как и предыдущий метод, хромосомный микроматричный анализ имеет некоторые ограничения. Он не позволяет выявлять или ограничен в выявлении таких аномалий, как:
- сбалансированные хромосомные аномалии, когда в хромосомах происходят изменения, которые не приводят к добавлению или утрате генетического материала. К ним относятся инверсии (разворот участка хромосомы на 180 градусов), реципрокные транслокации (обмен участками между хромосомами), небольшие инсерции (вставки в хромосомах);
- мозаицизм, если клеток с нарушениями в кариотипе менее 15%;
- CNV (copy number variation) – повторы небольших участков генома;
- точечные мутации – замены отдельных «букв» генетического кода;
- экспансия (увеличение) повторов коротких участков в ДНК;
- аномалии метилирования – присоединения особых метильных групп к определенным участкам ДНК, которые меняют активность генов.
Мутации в генах и заболевания, к которым они способны приводить
Мутации – это изменения, которые происходят в ДНК как случайным образом, так и под действием разных факторов, например химических веществ, ионизирующих излучений. Они могут затрагивать как отдельные «буквы» генетического кода, так и большие участки генома. Мутации происходят постоянно, и это основной двигатель эволюции. Чаще всего они бывают нейтральными, то есть ни на что не влияют, не приносят ни вреда, ни пользы. В редких случаях встречаются полезные мутации – они дают организму некоторые преимущества. Также встречаются вредные мутации – из-за них нарушается работа важных белков, наоборот, происходят достаточно часто. Генетические изменения, которые происходят более чем у 1% людей, называются полиморфизмами – это нормальная, естественная изменчивость ДНК Полиморфизмы ответственны за множество нормальных отличий между людьми, таких как цвет глаз, волос и группа крови.
Все внешние признаки и особенности работы организма, которые человек получает от родителей, передаются с помощью генов. Это важнейшее свойство всех живых организмов называется наследственностью. В зависимости от того, как проявляются гены в тех или иных признаках, их делят на две большие группы.
- Доминантные гены. Выражаясь простым языком, эти гены более «сильные». Если в клетках присутствует хотя бы одна копия такого гена, его признаки проявятся.
- Рецессивные гены «слабее» доминантных. Если у человека одна копия гена доминантная и одна рецессивная, – проявится признак доминантной. А для проявления рецессивного признака нужно две соответствующих копий.
Например, карий цвет глаз у человека является доминантным. Поэтому у кареглазых родителей с высокой вероятностью родится кареглазый ребенок. Если у одного из родителей глаза карие, а у другого голубые, то вероятность рождения кареглазых детей в такой семье тоже высока. У двух голубоглазых родителей, скорее всего, все дети тоже будут голубоглазыми. А вот у кареглазых родителей может родиться ребенок с голубыми глазами, если у обоих есть рецессивные «гены голубоглазости», и они достанутся ребенку. Конечно, это упрощенная схема, потому что за цвет глаз отвечает не один, а несколько генов, но на практике эти законы наследования зачастую работают. Аналогичным образом потомству могут передаваться и наследственные заболевания. 
Как выявляют рецессивные мутации?
Для выявления мутаций, которые передаются рецессивно, используют целый ряд исследований.
Секвенирование по Сэнгеру – метод секвенирования (определения последовательности нуклеотидов, буквально – «прочтение» генетического кода) ДНК, также известен как метод обрыва цепи. Анализ используется для подтверждения выявленных мутаций. Это лучший метод для идентификации коротких тандемных повторов и секвенирования отдельных генов. Метод может обрабатывать только относительно короткие последовательности ДНК (до 300–1000 пар оснований) одновременно. Однако самым большим недостатком этого метода является большое количество времени, которое требуется для его проведения.
Если неизвестно, какую нужно выявить мутацию, то используют специальные панели.
Панель исследования — тестирование на наличие определенных мутаций, входящих в перечень конкретной панели исследования. Анализ позволяет выявить одномоментно разные мутации, которые могут приводить к генетическим заболеваниям. Анализ позволяет компоновать мутации в панели по частоте встречаемости (скрининговые панели, направленные на выявление носительства патологической мутации, часто встречаемой в данном регионе или в определенной замкнутой популяции) и по поражаемому органу или системе органов (панель «Патология соединительной ткани»). Но и у этого анализа есть ограничения. Анализ не позволяет выявить хромосомные аберрации, мозаицизм и мутации, не включенные в панель, митохондриальные заболевания, а также эпигенетические нарушения.
Не в каждой семье можно отследить все возможные рецессивные заболевания. Тогда на помощь приходит секвенирование экзома – тест для определения генетических повреждений (мутаций) в ДНК путем исследования в одном тесте практически всех областей генома, кодирующих белки, изменения которых являются причиной наследственных болезней.
Секвенирование следующего поколения-NGS – определение последовательности нуклеотидов в геномной ДНК или в совокупности информационных РНК (транскриптоме) путем амплификации (копирования) множества коротких участков генов. Это разнообразие генных фрагментов в итоге покрывает всю совокупность целевых генов или, при необходимости, весь геном.
Анализ позволяет выявить точечные мутации, вставки, делеции, инверсии и перестановки в экзоме. Анализ не позволяет выявить большие перестройки; мутации с изменением числа копий (CNV); мутации, вовлеченные в трехаллельное наследование; мутации митохондриального генома; эпигенетические эффекты; большие тринуклеотидные повторы; рецессивные мутации, связанные с Х-хромосомой, у женщин при заболеваниях, связанных с неравномерной Х-деактивацией, фенокопии и однородительские дисомии, и гены, имеющие близкие по структуре псевдогены, могут не распознаваться. 
Что делать, если в семье есть наследственное заболевание?
Существуют два способа выявить наследственные генетические мутации у эмбриона:
Предимплантационное генетическое тестирование (ПГТ) в цикле ЭКО. Это диагностика генетических заболеваний у эмбриона человека перед имплантацией в слизистую оболочку матки, то есть до начала беременности. Обычно для анализа проводится биопсия одного бластомера (клетки зародыша) у эмбриона на стадии дробления (4–10 бластомеров). Существует несколько видов ПГТ: на хромосомные отклонения, на моногенные заболевания и на структурные хромосомные перестройки. Данные Simon с соавторами (2018) говорят о том, что в случае проведения ЭКО с ПГТ у пациентки 38–40 лет результативность ЭКО составляет 60%. Но при исследовании эмбриона есть ряд ограничений. Так, из-за ограниченного числа клеток можно не определить мозаицизм.
Если нет возможности провести ЭКО с ПГТ, то используют второй вариант – исследование плодного материала во время беременности.
Для забора плодного материала используют инвазивные методы:
- биопсия хориона – когда берут клетки из плаценты;
- амниоцентез – когда берут клетки амниотической жидкости.
Далее эти клетки исследуют при помощи одного или нескольких генетических тестов (которые имеют свои ограничения). Проведение инвазивных методов может быть связано с риском для беременности порядка 1%.
Таким образом, проведя дополнительные исследования, можно значительно снизить риск рождения ребенка с генетическим заболеванием в конкретной семье. Но привести этот риск к нулю на сегодняшний день, к сожалению, невозможно, так как любой генетический тест имеет ряд ограничений, что делает невозможным исключить абсолютно все генетические болезни.

Автор статьи
Пелина Ангелина Георгиевна
Ведёт генетическое обследование доноров Репробанка, осуществляет подбор доноров для пар, имеющих ранее рождённых детей с установленной генетической патологией.
Источник


