Аномалия Арнольда-Киари I
Аномалия Арнольда-Киари I — опущение миндалин мозжечка в большое затылочное отверстие со сдавливанием продолговатого мозга. Может сочетаться с сирингомиелией, базилярной импрессией или инвагинацией, ассимиляцией атланта. Симптомы коррелируют от степени снижения.
Эпидемиология
Аномалия Арнольда-Киари I встречается чаще у женщин [2].
Клиническая картина
В отличие от пороков развития Киари II, III и IV, аномалия Арнольда Киари I часто остается бессимптомной.
Вероятность клинических проявлений пропорциональна степени опускания миндалин. Все пациенты с пролабированием миндалин больше 12 мм имеют какую-либо симптоматику тогда, как примерно в 30% пролабирование в диапазоне между 5 и 10 мм протекают бессимптомно [1].
Компрессия ствола мозга (продолговатого мозга) может вызывать сирингомиелию с соответствующими симптомами и клинической картиной (затылочные боли, нарушение глотания, атаксия) разной выраженности, симптомами поражения спинного мозга и др.
Сопутствующие заболевания
Сирингомиелия шейного отдела позвоночника встречается в
35% (варьирует от 20 до 56%), гидроцефалиия в 30% [1,3] случаев, в обоих случаях считается данные изменения развиваются в результате нарушения ликовродинамики, центральном канале и вокруг спинного мозга.
В
35% (23-45%) выявляются скелетные аномалии [1, 3]:
- платибазия/ базилярная импрессия
- атланто-затылочная ассимиляция
- деформация Шпренгеля (Sprengel)
- синдром Клиппель-Фейля (Klippel-Feil)
Патология
Аномалия Арнольда Киари I характеризуется пролабированием миндалин мозжечка через большое затылочное отверстие, в основном в результате несоответствия между размерами мозжечка и задней черепной ямке. Аномалию Арнольда Киари I следует отличать от эктопии миндалин, которая протекает бессимптомно и является случайной находкой, при которой, миндалины выступают через затылочное отверстие не более чем на 3-5 мм 2.
Патология подтверждается при измерения максимального расстояния, на которое миндалины выступают ниже плоскости большого затылочного отверстия (условной линии между ophisthion и basion (1) Базион — передний край большого затылочного отверстия. 2) Опистион — задний край большого затылочного отверстия)).
Значения используемые для постановки диагноза отличаются у разных авторов [2]:
- выше затылочного отверстия: норма
- 6 мм: аномалия Арнольда Киари 1
Некоторые авторы используют более простую градацию [1]:
- выше затылочного отверстия: норма
- 5 мм: аномалия Арнольда Киари 1
Положение миндалин мозжечка меняется с возрастом. У новорожденных миндалины расположены чуть ниже большого затылочного отверстия и спускаются ниже с ростом ребенка, достигая своей низшей точки в возрасте 5 — 15 лет. В дальнейшем они поднимаются на уровень большого затылочного отверстия [3]. Таким образом, снижение миндалин на 5 мм у ребенка будет скорее всего нормой, а во взрослом возрасте при данных изменениях следует подозревать патологию [3].
КТ
Современное объемное сканирование с высоким качеством сагиттальной реформации относительно хорошо визуализирует затылочное отверстие и миндалины, хотя отсутствие контрастности (по сравнению с МРТ) с трудом позволяет провести точную оценку. Чаще патология может быть заподозрена на аксиальных изображениях, когда мозговое вещество охватывает миндалины а спинномозговая жидкость представлена в малом количестве или отсутствует. Данное состояние называется, в переполнением затылочного отверстия.
МРТ
МРТ исследование является методом выбора так, как помимо лучшей визуализации атомический изменений есть возможность оценить ликвородинамику. Сагиттальные срезы наиболее оптимальны для оценки аномалии Арнольда Киари I. Аксиальные изображения так же дают картину «переполненого» затылочного отверстия.
Лечение и прогноз
Оперативное лечение применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев. Оно состоит в ламинэктомии (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки.
Источник
Синдром Арнольда — Киар
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Мальформация Арнольда — Киари (МАК) — это патология развития ромбовидного мозга: продолговатого и заднего мозга, в последний входит Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг из‑за небольших размеров опускаются ниже большого затылочного отверстия, что приводит к их ущемлению и нарушению ликвородинамики. МАК относят к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 наблюдений на 100 000 населения, а у новорожденных — 1 на 4–6 тысяч. Сегодня понятно, что распространенность синдрома Арнольда — Киари значительно больше. Из-за бессимптомного течения и в результате учета разных типов МАК цифры очень разнятся — от 14 до 30 %.
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835–1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851–1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835–1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Клеланд полагал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 г. канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891–1976 гг.) и его коллега предложили «теорию тяги»: в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В «унифицированной» теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования говорят о том, что существуют разные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без. Описаны семейные случаи МАК 2 типа, однако роль генетических факторов еще недостаточно изучена.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20–40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. МАК 2 типа протекает более тяжело, дети с такой патологией уже рождаются с гидроцефальной формой черепа. Гидроцефалия препятствует нормальному развитию. Кроме того, такие дети страдают нарушениями дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Выраженность неврологической симптоматики в первую очередь зависит от выраженности нарушений ликвородинамики, а не от степени эктопии миндалин мозжечка.
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Пациенты, имеющие мальформацию Арнольда-Киари 0 и 1 типа, могут даже не знать о наличии у себя этого заболевания в течение всей жизни. Вследствие пренатальной диагностики МАК II, III и IV типа дети с данной патологией рождаются всё реже, а современные технологии выхаживания позволяют значительно увеличить продолжительность жизни таких детей.
Источник
Аномалия Киари ( Синдром Арнольда-Киари )
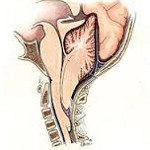
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
МКБ-10
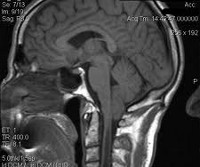
Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
- Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
- Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
- Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
- Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Источник


