Делеция гена что это значит
Мутации могут также вызываться инсерцией, инверсией, слиянием или делецией последовательности ДНК. Некоторые делеции и инсерции захватывают только несколько нуклеотидов и обычно легко обнаруживаются при секвенировании. В других случаях утрачивается, инвертируется, дублируется или транслоцируется значимый сегмент или целый ген, создавая новое размещение генной последовательности.
Такие мутации обычно обнаруживают на уровне блот-гибридизации ДНК пациента по Саузерну или ПЦР, выявляющими новые последовательности, созданные транслоцированным сегментом. В редких случаях делеции достаточно велики, чтобы быть видимыми на цитогенетическом уровне. Чтобы обнаруживаться даже с помощью высокоразрешающих методов окраски на стадии прометафазы, эти мутации обычно должны захватывать от 2 до 4 миллионов пар оснований ДНК.
Во многих примерах такие делеции захватывают более одного гена и вызывают синдромы генных последовательностей. Межхромосомные транслокации легче всего обнаружить методом SKY.
Небольшие делеции и инсерции
Некоторые делеции и инсерции влияют на незначительное число пар оснований. Когда число пар не кратно трем (т.е. захватывает не целое количество кодонов) и мутация находится в кодирующей последовательности, изменяется рамка считывания, начиная с участка инсерции или делеции. Такие мутации называются мутациями сдвига рамки.
В точке инсерции или делеции генерируется другая последовательность кодонов, кодирующая несколько аномальных аминокислот и завершающаяся стоп-кодоном в смещенной рамке. Если число оснований кратно трем, сдвига рамки не происходит, и в продукте гена обнаруживается инсерция или делеция соответствующих аминокислот.
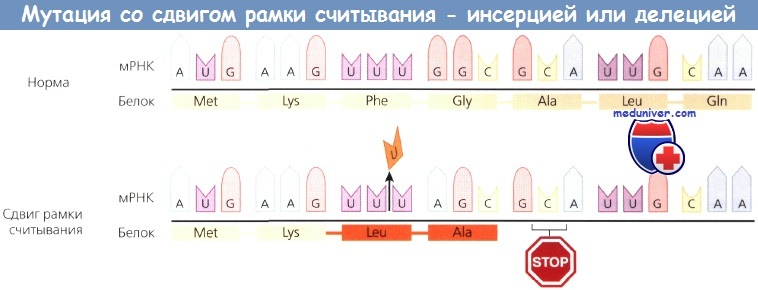 Мутация со сдвигом рамки считывания, при которой вставка (инсерция) или выпадение (делеция) пары оснований ведут к образованию стоп-кодона и, как правило, синтезу укороченного белка
Мутация со сдвигом рамки считывания, при которой вставка (инсерция) или выпадение (делеция) пары оснований ведут к образованию стоп-кодона и, как правило, синтезу укороченного белка
Большие делеции и инсерции
Изменения структурных генов, достаточно большие, чтобы выявляться блот-гибридизацией по Саузерну, встречаются сравнительно редко, но описаны при многих наследственных болезнях. Частота таких мутаций заметно отличается среди разных генетических заболеваний; некоторые характеризуются высокой частотой обнаруживаемых делеции, при других делеции бывают редкой причиной мутации.
Например, делеции большого гена дистрофина в Х-хромосоме при мышечной дистрофии Дюшенна или большого гена нейрофибромина при нейрофиброматозе I типа присутствуют более чем в 60% случаев. Большинство случаев а-талассемии — следствие делеции одного из двух генов b-глобина в хромосоме 16, тогда как b-талассемия только изредка вызывается делениями в гене b-глобина.
В некоторых случаях основа делеции гена хорошо известна и связана с нарушением рекомбинации между многочисленными копиями аналогичных или идентичных последовательностей ДНК. В других случаях причины делеции неизвестны.
Инсерция больших участков ДНК — причина мутаций, встречающихся значительно реже делеции. Тем не менее новый механизм мутации, инсерция последовательности LINE, описан в нескольких не родственных спорадических случаях у больных с гемофилией.
Семейство повторных последовательностей LINE представляет класс повторяющейся ДНК, которая может транскрибироваться в РНК, которая, в свою очередь, после обратной транскрипции генерирует последовательности ДНК, способные включать себя в различные места в геноме. У нескольких пациентов с гемофилией А в экзоне гена фактора VIII обнаружена прерывающая кодирующую последовательность и инактивирующая ген LINE последовательность длиной несколько килобаз.
Это обнаружение позволяет предполагать, что по крайней мере некоторые из 850 000 копий семейства LINE, имеющихся в геноме человека, способны вызвать болезнь за счет инсерции.
Эффекты рекомбинации
Важная причина мутаций при некоторых заболеваниях — делеции или дупликации, вызванные рекомбинацией весьма похожих или идентичных последовательностей ДНК. Например, как причина дупликации нескольких экзонов при семейной гиперхолестеринемии подтверждена рекомбинация между разными участками семейства диспергированных повторов класса Alu, располагающихся в интронах гена рецептора ЛПНП.
В других случаях ген может принадлежать семейству генов, представленных аналогичными копиями гена, располагающихся в хромосоме последовательно. Когда члены такого семейства генов располагаются тандемно в одном хромосомном регионе, они иногда неправильно спариваются или в мейозе (когда спариваются два гомолога), или после репликации в митозе (когда обмениваются ДНК две сестринские хроматиды).
Рекомбинация, происходящая между неправильно спаренными хромосомами или сестринскими хроматидами, может привести к делеции или дупликации гена. Считают, что неравный кроссинговер вызывает делецию одного из генов b-глобина при а-талассемии и изменения в числе копий гена зеленого пигмента в кластере генов красного и зеленого зрительного пигмента в Х-хромосоме как у людей с нормальным цветовым зрением, так и у мужчин со Х-сцепленными дефектами восприятия красного и зеленого цветов.
Возможны также аномальные спаривание и рекомбинация между двумя сходными последовательностями, повторяющимися в одной нити ДНК; в зависимости от ориентации последовательностей, такая рекомбинация может вести к делеции или инверсии. Например, почти половина всех случаев тяжелой гемофилии — следствие рекомбинации, инвертирующей множество экзонов и нарушающей структуру гена.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Делеция гена что это значит
Определение хромосомных мутаций в генах SMN1 и SMN2, являющихся причиной развития спинальной мышечной атрофии. В состав исследования входит исследование экзонов (участков) 7 и 8 гена SMN1 и определение количества копий гена SMN2.
- Экзон 7 гена SMN1
- Экзон 8 гена SMN1
- Определение количества копий гена SMN2
Спинальная мышечная атрофия, SMN ген (ген выживаемости моторных нейронов), генетическое обследование.
Синонимы английские
Spinal Muscular Atrophy, gene SMN (survival motor neuron gene), genetic research.
Локализация гена на хромосоме
Общая информация об исследовании
Спинальная мышечная атрофия является нервно-мышечным заболеванием, наследуется по аутосомно-рецессивному типу. Вследствие генетических изменений происходит разрушение двигательных нейронов, что ведет к ослаблению функции скелетных мышц, а впоследствии к их полной атрофии. Чаще всего патологический процесс происходит в мышцах нижних конечностей, головы, шеи. Различаются несколько типов спинальной мышечной атрофии в зависимости от возраста, в котором началось заболевание, и степени тяжести клинических проявлений.
Причиной возникновения спинальной мышечной атрофии является генетическая мутация. Ген SMN (survival of motor neuron protein) отвечает за развитие и функционирование белка двигательных нейронов спинного мозга. Существуют два подвида гена SMN: SMN1 и SMN2. Особо важным участком гена SMN1 является экзон 7, кодирующий информацию о белке, который входит в состав моторных нейронов. Также за данный белок отвечает экзон 8 гена SMN1. Различные нарушения в этих фрагментах ведут к изменению структуры белка и могут привести к развитию спинальной мышечной атрофии. Ген SMN2 практически идентичен гену SMN1, они отличаются лишь определенным нуклеотидом, который расположен в 7-м экзоне. Если происходит изменение в данном участке гена SMN2, то это также приводит к синтезу функционально дефектного белка. В то же время с гена SMN2 возможен синтез частично правильно работающего белка, что делает возможным сохранение некоторой функции двигательных нейронов. Поэтому количество копий гена SMN2 может влиять на степень клинических проявлений заболевания – если количество этих копий достаточно, то возможно более легкое течение заболевания.
Генетическая диагностика осложнена очень высокой степенью схожести (до 99%) генов SMN1 и SMN2. Наиболее значимую роль играет тщательное изучение экзона 7 и 8 гена SMN1, а также количества копий гена SMN2 (т.к. в них есть участки, отвечающие за возможность синтеза функционально полноценного белка). Для диагностики может быть использована полимеразная цепная реакция (ПЦР), которая позволяет получить большое количество копий нужного гена с помощью специальных реактивов. После получения нужного количества копий гена путем последовательного проведения сложных реакций выявляют правильность последовательности нуклеотидов в интересующем участке ДНК гена (для гена SMN1 это экзоны 7 и 8). Вследствие этого можно обнаружить точечные мутации или делецию (отсутствие) в тех или иных участках хромосом. Также оценивается ген SMN2 путем подсчета количества его копий, т.к. это имеет большое значение для прогноза степени тяжести клинических проявлений.
После проведения данных реакций и оценки наличия генетических мутаций можно делать выводы о наличии или отсутствии спинальной мышечной атрофии или предпосылок к ее развитию. При гомозиготном типе наследования генетические мутации обнаруживаются в обеих хромосомах, несущих ген спинальной мышечной атрофии (по одной от каждого из родителей), и тогда заболевание диагностируется. При гетерозиготном типе наследования мутация определяется только на одной хромосоме, что не проявляется заболеванием, но означает носительство генетической мутации и может вести к развитию патологии у детей данного лица.
Для чего используется исследование?
- Для верификации диагноза «спинальная мышечная атрофия» при наличии у пациента характерной клинической симптоматики;
- для оценки степени тяжести клинических проявлений и прогноза спинальной мышечной атрофии;
- для оценки риска рождения ребенка со спинальной мышечной атрофией или носительством ее генетической мутации.
Когда назначается исследование?
- При подозрении на спинальную мышечную атрофию на основе анамнеза и клинической симптоматики;
- при дифференциальной диагностике болезни моторных нейронов;
- при дифференциальной диагностике мышечной слабости;
- при раннем выявлении заболевания у родственников;
- при планировании семьи — для оценки генетического риска для будущего ребенка.
Что означают результаты?
Экзон 7 гена SMN1
Обнаружено 2 копии 7 экзона
гена SMN1
Экзон 8 гена SMN1
Обнаружено 2 копии 8 экзона
гена SMN1
Определение количества копий гена SMN2
Количество копий гена SMN2 — 1
копия гена; Количество копий
гена SMN2 — 2 копии гена;
Количество копий гена SMN2 — 3
копии гена; Количество копий
гена SMN2 — 4 копии гена
Если мутация определяется в обеих копиях гена (т.е. в обеих хромосомах), то это ведет к развитию спинальной мышечной атрофии.
Обнаружение нарушения в одной хромосоме свидетельствует о носительстве, заболевание при этом не возникает, но у двух носителей существует риск рождения детей с патологией.
Отсутствие мутации в обеих копиях гена свидетельствует об отсутствии риска развития спинальной мышечной атрофии и у самого человека, и у детей.
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, врач-генетик, врач-репродуктолог.
- Walter MC, Stauber AJ. Spinal muscular atrophy — clinical spectrum and therapy. Fortschr Neurol Psychiatr. 2018 Sep;86(9):543-550.
- Tizzano EF, Zafeiriou D. Prenatal aspects in spinal muscular atrophy: From early detection to early presymptomatic intervention. Eur J Paediatr Neurol. 2018 Sep 3.
- Harahap NIF, Niba ETE, Ar Rochmah M, Wijaya YOS, Saito T, Saito K, Awano H, Morioka I, Iijima K, Lai PS, Matsuo M, Nishio H, Shinohara M. Intron-retained transcripts of the spinal muscular atrophy genes, SMN1 and SMN2. Brain Dev. 2018 Sep; 40(8):670-677.
- Arnold ES, Fischbeck KH. Spinal muscular atrophy. Handb Clin Neurol. 2018; 148:591-601.
- Wu S, Li YL, Cheng NY, Wang C, Dong EL, Lu YQ, Li JJ, Guo XX, Lin X, Lai LL, Liu ZW, Wang N, Chen WJ. c.835-5T>G Variant in SMN1 Gene Causes Transcript Exclusion of Exon 7 and Spinal Muscular Atrophy. J Mol Neurosci. 2018 Jun; 65(2):196-202.
Источник
Что такое делеция, структурные хромосомные мутации?
Статья опубликована: 2018-12-25
Рейтинг: 5 из 5

Геном человека устроен таким образом, что для нормальной жизни у нас должно быть ровно 23 пары хромосом. Хромосомы существуют именно парно, потому что одна хромосома в паре приходит от мамы, а другая от папы. Нарушение в числе хромосом приводит к тяжелым заболеваниям, например, синдрому Дауна. Однако, количество хромосом может быть в норме, но все равно человек имеет аномалии в развитии, которые не совместимы с нормальной и долгой жизнью.
Дело в том, что могут быть отклонение в самой структуре хромосомы. Из хромосомы выпадают участки. Такое явление называется делецией. Делеция происходит от лат. deletio что значит уничтожение. Такая аномалия может возникать в результате неравного кроссинговера или при разрыве хромосомы.
Признаки делеционного синдрома
Делеции или микроделеции могут вообще внешне не проявляться. Человек может всю жизнь прожить с этим синдромом и не знать о его существовании. Заболевание может обнаружится, например, когда возникнет необходимость завести ребенка, но попытки будут безуспешными.
Существуют делеции с явно выраженными внешними признаками:
- Синдром Ди Джорджи. Ребенок с этим синдромом может иметь отклонения в лицевой области: расщепление неба, “заячья губа”, деформированные ушные раковины, маленький рот, широкую переносицу. Также часты пороки сердца, мочеполовой системы, умственные отклонения.
- Синдром делеции 1p36. К внешним лицевым признакам относятся: маленький рот, близко посаженные глаза, заостренный подбородок, маленькие уши, плоский нос, поздно закрывающийся родничок. Также наблюдается наличие аутизма и задержек в умственном развитии. Пороки сердечно-сосудистой системы. Нарушение развития речи и слуха.
- Синдром «кошачьего крика». Внешне проявляется так: микроцефалия, аномальный разрез глаз, недоразвитые хрящи ушных раковин, недоразвитие нижней челюсти. Характерным признаком является деформация гортани при которой плач ребенка похож на мяуканье голодного котенка (более высокие тона, чем у обычного плача).
Причины появления делециий
Существует несколько причин, при которых возможно развитие делеционного синдрома:
- Наследственные хромосомные аномалии, полученные от родителей;
- Возраст матери более 40 лет и отца болен 45 лет;
- Перенесение тяжелых заболеваний на ранних сроках беременности и при зачатии;
- Плохой анамнез и т.д.
Диагностика синдромов
Выявить наличие делеций возможно на ранних сроках беременности. Уже на 9-ой недели по венозной крови матери можно провести ДНК анализ и диагностировать заболевание. Такое генетическое исследование называется неинвазивный пренатальный ДНК тест. Тест проводится без направления врача, т. к. не имеет противопоказаний и побочных эффектов.
Генетический центр “ДТЛ” выполняет НИПТ тесты уже более 4 лет. В сотрудничестве с ведущими мировыми лабораториями в области пренатальной диагностики нами проведено уже болен 1000 подобных исследований.
Источник


