- Расшифровка анализа на кариотипирование
- Что значит изменение количества хромосом в кариотипе
- Таблица расшифровки аномалий половых хромосом
- Инверсия пола 46 XY
- Кариотипирование супругов. Анализ на кариотип
- Кариотип и кариотипирование супругов
- Как сдать анализ на кариотип. Подготовка.
- Хромосомный микроматричный анализ (ХМА)
Расшифровка анализа на кариотипирование
Нормальный кариотип человека содержит 46 хромосом: 22 пары аутосом и 1 пару половых хромосом – ХХ у женщин и ХY у мужчин. Нарушение этой генетической структуры является причиной наследственных болезней, бесплодия и самопроизвольного прерывания беременности.
Плохой результат кариотипирования выглядит как:
- Изменение общего числа хромосом (анеуплоидия).
- Аномалии их структуры (абберации) – удвоение или потеря участка хромосомы, накладка одной части на другую, изменение порядка генов из-за перекрута на 180 градусов.
Что значит изменение количества хромосом в кариотипе
Аномалии кариотипа приводят к порокам развития плода, поэтому такие беременности в 50-60% случаев заканчиваются выкидышем в 1 триместре. Избыток генетического материала – причина трисомий 13 (47ХХ или ХY, 13+), 18 (47ХХ или XY, 18+) и синдрома Дауна (47ХХ или ХY, 21+).
Потеря участка короткого плеча пятой хромосомы (46ХХ или ХY,5р-) вызывает синдром кошачьего крика, такой же дефект длинного плеча пятнадцатой хромосомы (46ХХ или ХY, del15q11-q13) – болезнь Прадера Вилли, признаками которой являются низкие тонус мышц и плотность костей, задержка психического и речевого развития, гипогонадизм.
Нарушения кариотипа, связанные с гоносомами, имеют менее выраженные патологические проявления: их носители могут отличаться только высоким ростом и чуть сниженным интеллектом.
Таблица расшифровки аномалий половых хромосом
| Кариотип | Заболевания |
|---|---|
| 45ХО | Синдром Шерешевского-Тернера, встречается только у женщин (1:2000 случаев). Для синдрома Тернера характерны бесплодие, рост на 20-30 см ниже среднего, короткая шея, низкое положение ушных раковин. |
| 47ХХХ | Синдром трисомии-Х, чаще всего определяется случайно, при профилактическом обследовании. Это здоровые внешне женщины с генетической склонностью к невынашиванию беременности и хромосомным аномалиям у потомков. |
| 47ХYY | Синдром полисомии-Y встречается у мужчин высокого роста (1:1000 случаев), способных зачать ребенка. Для носителей полисомии-Y характерны сниженный интеллект и склонность к агрессии. |
| 47ХХY | Синдром Клайнфельтера – мужская патология (1:500 случаев), может проявляться в виде азооспермии, отсутствия сперматозоидов в эякуляте и бесплодия. |
Результаты кариотипирования помогут доктору выбрать тактику ведения бесплодной пары или пациентки с привычным невынашиванием беременности, а также оценить шансы супругов на здоровое потомство.
Также в МЖЦ у вас есть возможность сдать биоматериал (кровь или слюну) для детального ХМА-анализа, диагностирующего более 1000 генетических синдромов и болезней, связанных с аберрациями.
Источник
Инверсия пола 46 XY
OMIM 400044
Наша команда профессионалов ответит на ваши вопросы
Инверсия пола, 46,XY
Наличие женского фенотипа при нормальном мужском кариотипе характеризует XY-инверсию пола. Наиболее частой причиной данного нарушения формирования пола является синдром Свайера – это полная или «чистая» дисгенезия гонад при кариотипе 46,XY. Частота XY-дисгенезии гонад составляет 1 на 30000 человек. Больные имеют женский фенотип без признаков двойственности полового развития: феминное телосложение, развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку и маточные (фаллопиевы) трубы. Однако у пациентов с синдромом Свайера практически отсутствуют женские половые железы, которые в данном случае представлены дисгенетичными гонадами, представляющими собой соединительнотканные тяжи (стреки) с небольшими включениями железистой ткани, овариально-подобной стромы без фолликулов. Как правило, диагностирование синдрома Свайера происходит у девочек в пубертатный период, когда у них не происходит нормального полового развития. Причиной обращения к врачу при этом является задержка полового развития и отсутствие начала менструаций, реже наличие злокачественных новообразований, происходящих из дисгенетичных гонад. Так как дисгенетичные гонады подвержены озлокачествлению, показано их удаление в детстве или на момент постановки диагноза XY-дисгенезии гонад. После оперативного лечения пациенткам, как правило, еще в подростковом возрасте назначается заместительная гормональная терапия, чтобы достичь нормального развития вторичных половых признаков и предотвратить развитие остеопороза. У женщин с XY-дисгенезией гонад нет собственных яйцеклеток, однако в некоторых случаях она в состоянии выносить плод, полученный в программе ЭКО при оплодотворении донорской яйцеклетки сперматозоидами супруга.
Инверсия пола, 46,XY тип 1 (OMIM 400044)
Наиболее частой из известных причин «чистой» формы дисгенезии гонад 46,XY являются микроструктурные перестройки Y-хромосомы c утратой гена SRY (Sex-determining region Y), а также точковые мутации данного гена. У 10-15% больных с синдромом Свайера обнаруживают отсутствие локуса SRY. В большинстве случаев это обусловлено утратой фрагмента дистальной части короткого плеча Y-хромосомы (Yp11.3), вследствие X-Y транслокации. Еще у 10-15% пациентов с данным синдромом выявляют мутации гена SRY.
Ген SRY локализован на коротком плече Y хромосомы и кодирует транскрипционный фактор – белок, связывающийся с генами, определяющими развитие пола плода по мужскому типу. Мутации в гене SRY приводят к синтезу функционально неполноценного белка и к нарушению дифференцировки клеток Сертоли и формирования семенных канальцев в развивающихся бипотенциальных гонадах плода, что вызывает дисгенезию гонад и развитие остальных органов половой системы по женскому типу, несмотря на наличие Y-хромосомы в кариотипе.
Инверсия пола, 46,XYтип 2 (OMIM 300018)
Данный тип XY-инверсии пола обусловлен дупликаций гена NR0B1 (DAX-1). Ген NR0B1локализован на коротком плече Х хромосомы (локус Хp21.3). Кодируемый этим геном белок DAX-1 играет важную роль в развитии и функции некоторых органов эндокринной системы, в том числе и половых желез. Еще внутриутробно он контролирует активность генов, участвующих в формировании этих тканей, а в постнатальном периоде DAX-1 регулирует выработку в них гормонов. Белок DAX-1 оказывает дозо-зависимый эффект на органы эндокринной системы. Дупликация гена NR0B1, а также делеция располагающегося рядом с геном NR0B1 локуса, негативно-регулирующего его транскрипцию приводит к XY-инверсии пола, обусловленной XY-дисгенезией гонад часто сочетающейся с нарушением функции надпочечников. Точковые мутации этого гена у пациентов с кариотипом 46,XY вызывают нарушение развития тестикулярной ткани, приводят к дефициту маскулинизации. Мутации в этом гене также вызывают Х-сцепленную гипоплазию надпочечников, как у пациентов с кариотипом 46,ХХ так и 46,XY.
Инверсия пола, 46,XY тип 3 (OMIM 612965)
Данная форма XY-инверсии пола обусловлена мутациями гена NR5A1 (SF1). Ген NR5A1 кодирует транскрипционный фактор — стероидогенный фактор 1 (SF-1), с помощью которого контролируется активность ряда генов, кодирующих экспрессию белков-ферментов, ответственных за биосинтез стероидных гормонов в надпочечниках и гонадах, в том числе выработку половых гормонов. Функция белка SF-1 регулирует дифференцировку, развитие и функционирование, надпочечников, мужских и женских половых желез, сперматогенез и оогенез, развитие мужских или женских половых признаков. Ген NR5A1 локализуется на длинном плече хромосомы 9 (локус q33.3) и состоит из 7 экзонов (включая первый некодирующий экзон). Мутации в данном гене приводят к различным формам нарушения развития и функции половой и эндокринной систем. При этом нарушение дифференцировки и развития гонад, гаметогенеза может отмечаться как в сочетании, так и без поражения надпочечников (гипоплазия коры надпочечников). Помимо полной (характеризующейся наличием тяжевидных гонад при развитии остальных половых органов по женскому типу) и неполной формы (характеризующейся двойственным развитием гениталий) дисгенезии гонад 46,XY, мутации в гене NR5A1 могут приводить к развитию других заболеваний. Среди них: различная степень нарушения развития яичников у женщин с кариотипом 46,ХХ (от полной формы ХХ-дисгенезии гонад, неполной формы ХХ-дисгенезии гонад до синдрома преждевременной недостаточности яичников), развитие недостаточности коры надпочечников, синдрома тестикулярной дисгенезии и/или с нарушением сперматогенеза у 46,XY мужчин, бесплодие. Исследование на наличие герминальных мутаций в гене NR5A1 показано при различных нарушениях формирования пола, полового развития или репродуктивной функции, соматические мутации – при опухолях надпочечников. Важно отметить, что нарушения, вызванные мутациями данного гена, в отличие от других генов, могут иметь как аутосомно-доминантный, так и аутосомно-рецессивный тип наследования. При этом у пациентов, имеющих мутации гена NR5A1, может быть, как нормальный мужской 46,XY, так и нормальный женский 46,ХХ кариотип.
Инверсия пола, 46,XY тип 4 (OMIM 154230)
Эта форма XY-инверсии пола обусловлена делецией локуса 9p24.3. У пациенток отмечают нормально развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку, при гистологическом исследовании гонад обнаруживают наличие незрелой тестикулярной ткани, содержащей клетки Сертолли, и отсутствие зрелых половых клеток. Инверсия пола у данных пациентов, вероятно, обусловлена потерей одной из копий дозо-чувствительного гена, локализованного в данном локусе. Генами-кандидатами являются DMRT1 и DMRT2.
Инверсия пола, 46,XY тип 5 (OMIM 613080)
Данная аутосомно-рецессивная форма инверсии 46,XY обусловлена наличием мутаций в гене CBX2, расположенного на хромосоме 17 (локус 17q25). В 2009 году Байсон-Лаубер описал случай новорожденной девочки с кариотипом 46,XY, у которой в результате секвенированияв гене CBX2 были обнаружены две мутации (P98L и R443P). В результате исследований у девочки были обнаружены нормально развитые яичники, с наличием овариальной ткани и первичных фолликулов, а также влагалище и матка. Однако возраст еще был слишком мал, чтобы оценить ее фертильность и дальнейшее половое развитие.
Инверсия пола, 46,XY тип 6 (OMIM 613762)
XY-инверсия пола связана с наличием мутации в гетерозиготном состоянии в гене MAP3K1, расположенном в локусе 5q11.2. Пациентки с данной формой дисгенезии гонад имеют высокий рост, который, вероятно, обусловлен избыточной продукцией андрогенов, тяжевидные яичники, гипоплазированную матку, иногда наблюдается клиторомегалия.
Инверсия пола, 46,XY тип 7 (OMIM 233420)
Инверсия пола обусловлена наличием у пациенток мутаций в гомозиготном или компаунд-гетерозиготном состоянии в гене DHH, расположенного в локусе 12q13.12. У нескольких пациенток было описано наличие недоразвитой матки, также присутствовали фаллопиевы трубы и наблюдали полную форму ХY-дисгенезии гонад (тяжевидные гонады, которые часто озлокачествлялись).
Инверсия пола, 46,XYтип 8 (OMIM 614279)
Данный тип XY-инверсии пола обусловлен мутациями гена AKR1C2, лежащего в локусе 10p15, отвечающего за альтернативный путь синтеза дигидротестостерона. Мутации сцепленного с ним гена AKR1C4, который сегрегирует вместе с геном AKR1C2, могут влиять на выраженность фенотипических проявлений.
В Центре Молекулярной Генетики проводится молекулярный анализ ключевых генов, контролирующих дифференцировку пола, в частности выполняется секвенирование генов SRY и NR5A1 (SF1), а также с помощью количественного метода MLPA проводится поиск делеций и дупликаций генов SRY, NR5A1 (SF1), NR0B1 (DAX-1).
Источник
Кариотипирование супругов. Анализ на кариотип
Кариотип и кариотипирование супругов
Кариотипирование супругов – это углубленное лабораторное обследование для выявления изменений кариотипа, которое сдает пара в тех случаях, когда необходимо понять причину бесплодия, невынашивания беременности, а также заранее исключить генетические проблемы перед протоколом ЭКО и планированием беременности.
Кариотип – хромосомный набор человека с совокупностью признаков. Генетический фактор занимает достаточно большой процент среди супружеских пар с бесплодием, невынашиванием беременности, а также в группах мужчин с тяжелым нарушением сперматогенеза.
Анализ на кариотип сдают один раз в жизни. Поскольку это важный генетический анализ, то рекомендуется сдавать в специализированных центрах. В нашей лаборатории кариотипирование проводят высококвалифицированные лабораторные генетики. Анализируется материал всех 23 пар хромосом. Результат выдается согласно международной цитогенетической номенклатуре.
В норме результаты кариотипа выглядят следующим образом:
46, XX – нормальный женский кариотип;
46, XY – нормальный мужской кариотип.
Изменения кариотипа могут представлять собой изменения количества хромосом (анеуплоидии) либо структуры хромосом (или аберрации: транслокации, инверсии и др.). Внешне здоровый человек может быть носителем хромосомных аномалий, которые могут являться причиной бесплодия, невынашивания беременности или рождения у супружеских пар детей с пороками развития.

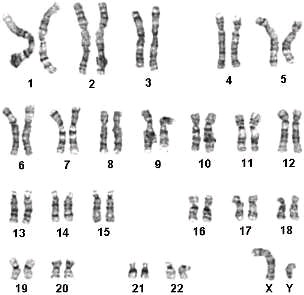
46, XX – нормальный женский кариотип 46, XY – нормальный мужской кариотип
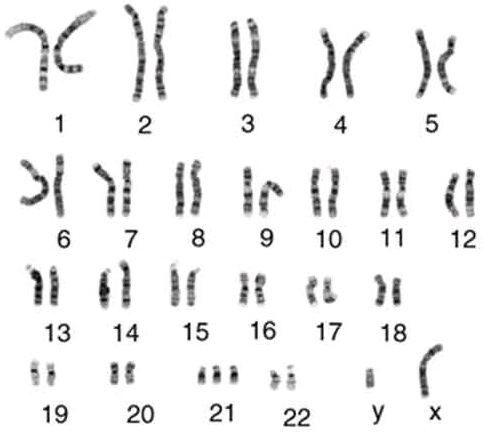
46, XY, +21 – дополнительная хромосома 21 (синдром Дауна)
- бесплодие в паре, в особенности наличие мужского фактора бесплодия;
- привычное невынашивание беременности;
- наличие выкидышей неясного генеза в анамнезе;
- наличие случаев анэмбрионии, рождения ребенка с множественными врожденными пороками развития (ВПР) или мертворождение;
- аменорея первичная или вторичная;
- наличие хромосомных аномалий у родственников супругов;
- планирование ЭКО;
- неудачные попытки ЭКО;
- прогноз здоровья для будущего потомства.
- Цитогенетический
- Молекулярно-генетический (Хромосомный микроматричный анализ)
Классическим методом определения кариотипа является цитогенетический. Также этим методом выполняют кариотипирование супругов. Метод основан на культивировании клеток крови с последующим приготовлением и фотографированием препаратов окрашенных хромосом. Метод ХМА представляет собой современную молекулярную технологию исследования кариотипа и показан при задержке развития и роста человека, наличием врожденных пороков развития (ВПР) у детей, аутизме, подозрении на микроделеционные синдромы.
В Лаборатории ЦИР кариотипирование супругов проводят цитогенетическим методом:
При кариотипировании без аберраций специалист анализирует под микроскопом 12-15 клеток. Анализ определяет количественные изменения числа хромосом. При этом будут обнаружены регулярные аберрации, которые встречаются в большом проценте клеток или во всех клетках. Обнаружение нерегулярных мутаций, которые являются следствием неблагоприятных факторов на организм (радиация, химические воздействия и др.) могут быть не обнаружены при таком количестве исследуемого материала.
При выполнении анализа кариотипа без аберраций (+фото) будет приложена фотография хромосом для наглядной оценки результата врачом-генетиком.
Кариотипирование с аберрациями — это расширенное генетическое обследование, при котором подробно анализируется 100 клеток, и рассчитывается процент аберраций. Исследование важно для супругов с бесплодием и невынашиванием беременности. Этот анализ способен выявлять возможные следы действия вредных факторов на геном человека. Из-за трудоемкости анализа (на исследование одного человека уходит целый рабочий день специалиста очень высокой квалификации) немногие медицинские центры делают анализ на аберрации. Заключение представляет собой результат кариотипа согласно международной цитогенетической номенклатуре хромосом человека вместе с указанием обнаруженных аберраций в определенной хромосоме.
Что такое аберрации?
Аберрации – изменения количества и структуры хромосом. Из количественных хромосомных аберраций различают анеуплоидии и полиплоидии. Из структурных:
- Инверсия – поворот участка хромосомы на 180 ○ ;
- Делеция – выпадение участка хромосомы;
- Транслокация – перенос части одной хромосомы на другую;
- Дупликация – удвоение участка хромосомы;
- И др.
Аберрации могут носить регулярный или нерегулярный характер. Регулярные аберрации обнаруживаются в большом проценте клеток или во всех клетках. Они возникают в момент зачатия или в первые дни после зачатия. Нерегулярные мутации чаще всего являются свидетельством действия на организм неблагоприятных факторов (радиация, химические вредности и пр.).
Почему экспертного уровня?
Мы являемся одной из немногих клиник, работающих более 20 лет в области репродукции. В нашей лаборатории цитогенетики кариотипирование проводится специалистами высокого уровня, где идет просмотр каждой хромосомы.
Как сдать анализ на кариотип. Подготовка.
Смотрите также:
«Замершая» беременность: каковы причины? Какое значение имеет генетический фактор в развитии «замершей» беременности?
Анализ на кариотип (кариотипирование). Как интерпретировать анализ на кариотип? Отвечает Гузов И.И.
ГЕНЫ и БЕРЕМЕННОСТЬ. Генетические причины невынашивания беременности.
Мозаичность кариотипа
Выявление мозаицизма кариотипа зависит от процентного содержания его (если есть) в кариотипе и от выбранной методики анализа. Однозначно при ХМА более вероятно обнаружение мозаицизма в пределах разрешающей способности (наличие более 25% в кариотипе). Также можно обнаружить мозаицизм при наличии его в исследуемых клетках в Определении кариотипа с аберрациями цитогенетическим методом. В связи с ограничениями цитогенетического метода если процент мозаицизма мал, то, скорее всего, его можно не увидеть.
Хромосомный микроматричный анализ (ХМА)
Однако для диагностики ряда заболеваний, связанных с хромосомными аномалиями, существует более современная технология исследования кариотипа – хромосомный микроматричный анализ. Анализ на кариотип выполняется молекулярно-генетическим методом aCGH (микроматричная сравнительная геномная гибридизация), который в отличие от классического цитогенетического метода, имеет высокую разрешающую способность, позволяющую обнаружить более мелкие структурные изменения кариотипа.
Этот анализ показан в следующих случаях:
- задержка психомоторного развития;
- задержка роста;
- задержка/нарушение полового созревания;
- аутизм и аутические расстройства;
- множественные ВПР (врожденные пороки развития);
- наследственные заболевания в семье;
- судорожные состояния;
- выяснение причин тяжелых осложнений беременности (потеря беременности, антенатальная гибель плода, мертворождение);
- подозрение на микроделеционные синдромы;
- при синдроме внезапной смерти ребенка.
ХМА проводится с использованием олигонуклеотидных микроматриц. Анализируется материал всех 23 пар хромосом. Разрешающая способность ХМА составляет 40 тыс. пар нуклеотидов по основной структуре и 20 тыс. пар нуклеотидов в таргетных областях, т.е. в тех зонах, где высока вероятность определить мутацию. Это и позволяет проводить исследование на гораздо большее количество синдромов, по сравнению с обычным цитогенетическим исследованием кариотипа. К ограничениям анализа относится невозможность выявления мозаицизма, полиплоидии, сбалансированных транслокаций, а также микроделеций и микродупликаций за границами разрешающей способности метода.
Методы диагностики хромосомной патологии
Источник


